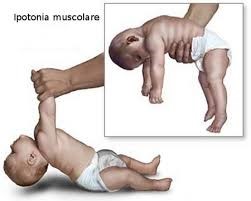
Le malattie neuromuscolari geneticamente determinate (ereditarie), quelle che colpiscono l’età infantile sono malattie ereditarie, ad evoluzione spesso progressiva, con interessamento più o meno diffuso della muscolatura scheletrica. Viene ad essere danneggiato uno dei componenti dell’unità motoria, che comprende il 2° motoneurone, placca neuromuscolare e muscolo.
Esordiscono in età infantile, con decorso rapido o possono comparire in età giovanile, con evoluzione più lenta. Difficile che si presentino in età avanzata.
La patologia neuromuscolare ereditaria sono:
- Neuropatia sensitivo-motoria periferiche: è la più frequente e colpisce una persona ogni mille;
- Distrofia Miotonica (DM);
- Distrofinopatie (Duchenne, DMD e Becker, BMD);
- Distrofia Muscolare dei 3 Cingoli;
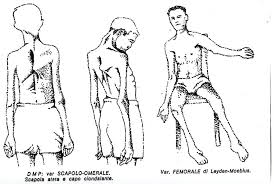
- Distrofia Muscolare Facio-Scapolo-Omerale (FSO);
- Atrofie Muscolari Spinali (SMA I, II, III);
- Miotonie Congenite.
L’obiettivo è quello di conservare il più allungo possibile l’autonomia, ritardare l’aggravamento con un buon intervento multifunzionale, verranno offerte tecniche, strategie e ausili compensative quando necessario.
Ritardare l’evoluzione della malattia è un obiettivo importantissimo; conservare il più a lungo possibile delle abilità, a volte fa sembrare l’intervento riduttivo, ma rappresenta un grande punto di forza per mantenersi funzionali.
Gli interventi su cui si va ad operare sono principalmente 4:
- funzioni motorie
- funzioni cognitivo-comunicativo
- funzioni di sopravvivenza
- sfera psicologica
Le quattro aree possono risultare compromesse e per questo devono essere analizzate ai fini di formulare una valutazione funzionale (diagnosi riabilitativa). Occorre valutare il disturbo, anche in termini quantitativi. Valutazioni periodiche e scadenzate, permetteranno di monitorare l’evoluzione della malattia, l’efficacia del trattamento e decidere eventuali modifiche da apportare
Personalmente ritengo molto importante l’ambiente sociale in cui vive il paziente; spesso si instaurano dei meccanismi non sempre corretti, dettati o da iperprotezione, per cui lo si va ad anticipare troppo nelle sue attività dinamiche, o al contrario, convinti di stimolarlo, lo “s’ignora” nelle sue difficoltà per attivare in lui una reazione risolutrice. La giusta via di mezzo, la consapevolezza reale di cosa può o non può fare in sicurezza deve essere rinforzata.
La fornitura di una carrozzina per gli spostamenti extradomiciliari deve avvenire quando la deambulazione autonoma è divenuta troppo faticosa e il rischio di cadute eccessivo: se tempestiva, viene spesso accolta dal bambino come una liberazione. Nella SMA il bisogno di ausili per gli spostamenti è più precoce e l’autonomia va perseguita con sollecitudine tenuto anche conto delle buone capacità intellettive di questi pazienti.
Gestione degli aspetti nutrizionali
E’ necessario l’intervento logopedico già nella fase iniziale della disfagia; si dovrà da subito avviare una riabilitazione della muscolatura fono-articolatoria e deglutitoria e nel caso avviare misure di sicurezza modificando la consistenza dei cibi (alimenti passati, a densità omogenea), utilizzo di addensanti e acque gelificate in caso di difficoltà con i liquidi.
Fondamentale istruire il paziente ed i familiari sulle tecniche di deglutizione (deglutizione sopraglottica) e sulle posture da assumere durante il pasto. (vedi disfagia e posture da adottare)
Quando la disfagia diventa severa, è indicata la nutrizione artificiale mediante la gastrostomia endoscopica percutanea (PEG). Altre modalità di nutrizione enterale sono il sondino naso-gastrico (SNG) e la gastrostomia inserita sotto guida radiologica (RIG).
Gestione dei disturbi respiratori
L’insufficienza respiratoria rappresenta la più frequente causa di morte nei pazienti affetti da SLA; normalmente si presenta nella fase avanzata della malattia, ma talora può evidenziarsi in fasi relativamente precoci.
Quali sono i segni e sintomi di una insufficienza respiratoria?
- Dispnea per minimo sforzo o nel parlare = noteremo Tachipnea
- Ortopnea utilizzo dei muscoli respiratori accessori
- Frequenti risvegli notturni = noteremo movimenti paradossi dell’addome
- Eccessiva sonnolenza diurna = noteremo ridotta espansione toracica
- Astenia = noteremo colpo di tosse debole
- Cefalea mattutina = noteremo sudorazione
- Difficoltà ad eliminare le secrezioni = noteremo tachicardia
- Apatia = noteremo confusione mattutina, allucinazioni
- Scarso appetito = noteremo perdita di peso
- Scarsa concentrazione e disturbi di memoria = noteremo secchezza delle fauci
Modalità di accesso al Centro e servizi offerti
La Regione Lazio ha individuato il Policlinico A. Gemelli come Centro di Riferimento per la SLA e l’Azienda Ospedaliera San Filippo Neri come Presidio per la SLA, e anche il policlinico Umberto I e l’Azienda Ospedaliera S Camillo.
I Centri sono autorizzati al rilascio della certificazione di malattia.
La SLA è inserita nell’elenco delle malattie rare ed è identificata dal codice di esenzione RF0100 (Allegato 1 del DM 279/2001 “Regolamento di istituzione della Rete Nazionale malattie rare e di esenzione dalla partecipazione al costo delle relative prestazioni sanitarie”, Allegato 2).
Dott.ssa Logopedista Claudia Antognozzi