La Sclerosi Laterale Amiotrofica (SLA)
La Sclerosi Laterale Amiotrofica (SLA) colpisce il neurone motorio (o motoneurone), cellula del sistema nervoso centrale. Abbiamo un’atrofia muscolare combinata ad un indurimento di alcune parti del midollo spinale.
Si tratta di una patologia degenerativa progressiva che comporta la perdita:
- dei motoneuroni superiori, situati nel cervello e nel tronco encefalico
- dei motoneuroni inferiori, situati nel tronco encefalico e nel midollo spinale.
Ciò porta alla perdita del controllo dei muscoli deputati al movimento e ad altre funzioni.
Circa il 10% dei pazienti con SLA presenta anche sintomi di demenza frontotemporale dovuta alla degenerazione dei neuroni di questa area del cervello.
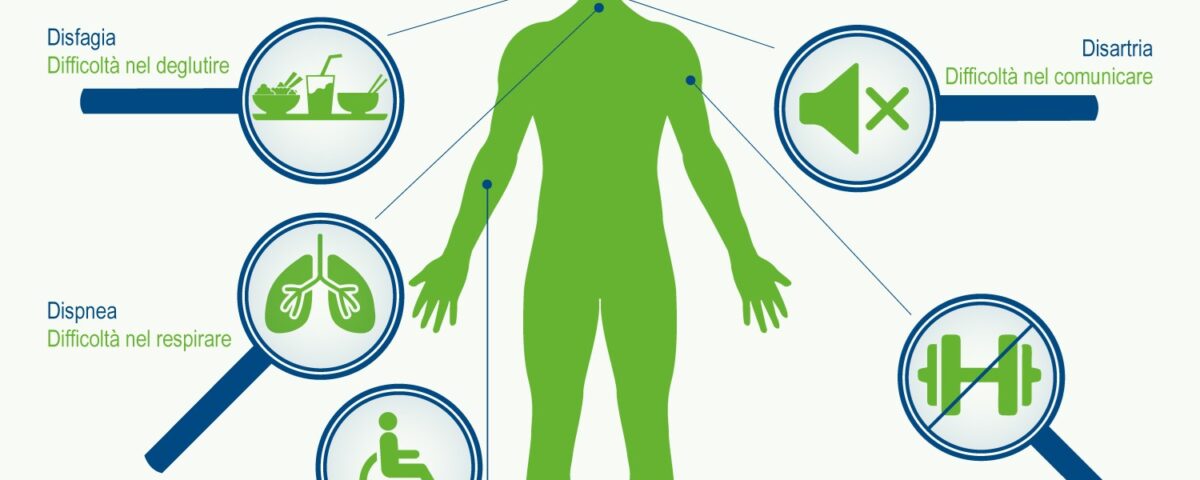
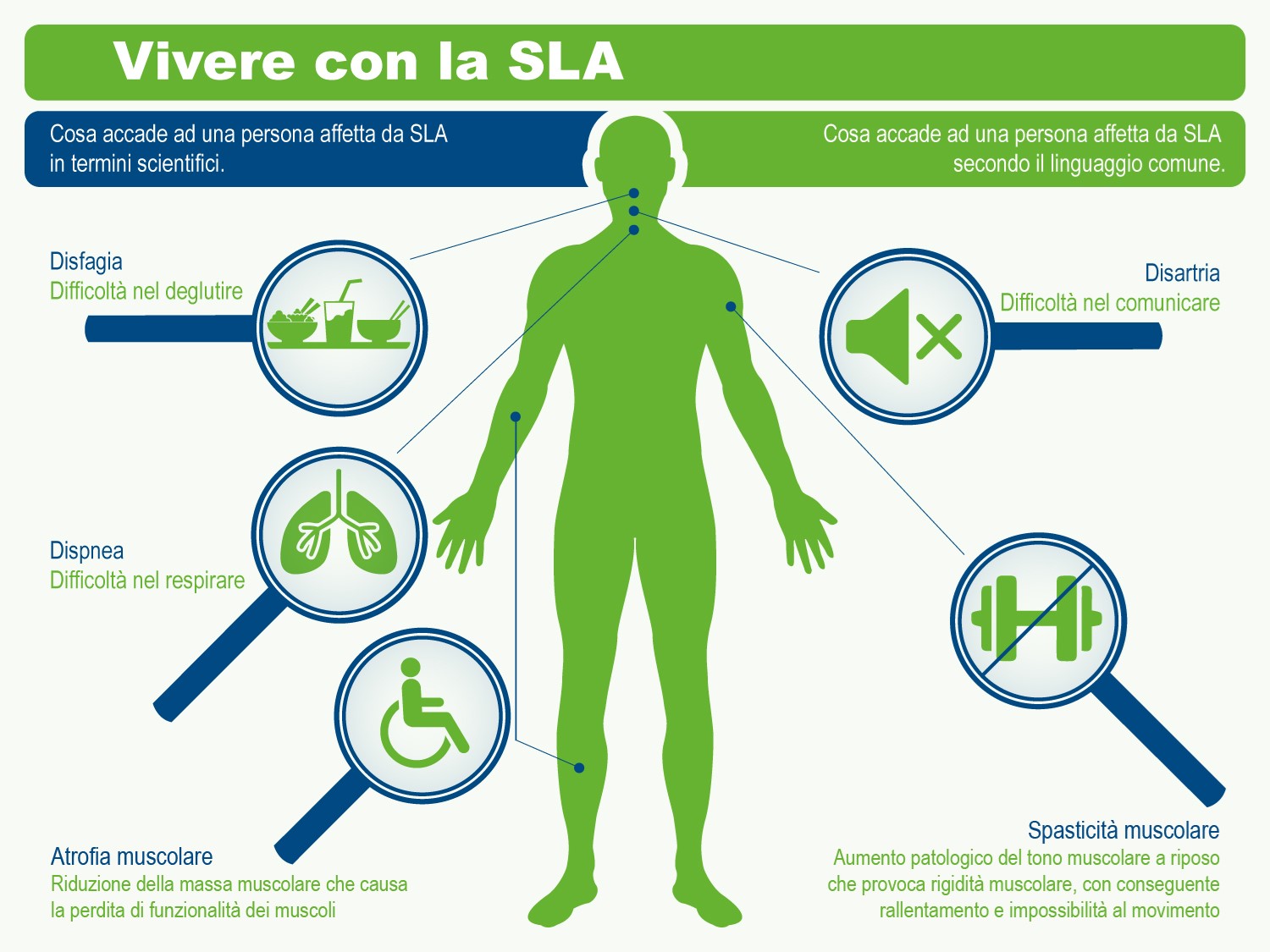
La malattia spesso inizia con spasmi muscolari e debolezza in un arto o difficoltà ad articolare la parola e progredisce sino a colpire tutti i muscoli necessari per muoversi, parlare, mangiare e respirare.
Sintomi
I primi sintomi della malattia possono passare inosservati.
Potrebbero manifestarsi inizialmente con delle brevi contrazioni e rigidità muscolare, debolezza dei muscoli fino ad arrivare ad alterato funzionamento di un braccio o di una gamba; la voce potrebbe risultare alterata.
Questi disturbi (debolezza e atrofia muscolare) piano piano saranno più e si inizierà a sospettare una forma di SLA quando ormai l’entità del danno sarà già considerevole (60-70% di motoneuroni).
I sintomi associati alla degenerazione dei motoneuroni superiori sono:
- aumento eccessivo del tono muscolare (ipertono muscolare)
- accentuazione esagerata dei riflessi muscolo-tendinei (iperreflessia profonda)
- risposta anomala al riflesso cutaneo plantare (segno di Babinski).
I sintomi associati alla perdita dei motoneuroni inferiori sono:
- riduzione del tono muscolare (ipotonia muscolare)
- riduzione del volume muscolare (atrofia muscolare)
- iporeflessia
- contrazione spontanea, rapida e regolare di uno o più muscoli, senza conseguente movimento (fascicolazioni).
Sintomi che si manifestano con il progredire della malattia sono:
- debolezza degli arti;
- crampi muscolari e fascicolazioni;
- difficoltà a camminare o a svolgere le normali attività quotidiane;
- difficoltà a masticare, a deglutire, a parlare e a respirare;
- cambiamenti delle funzioni cognitive e comportamentali.
Di norma il paziente non avverte dolore, tutti gli organi di senso rispondono adeguatamente agli stimoli; la sfera sessuale e il controllo sfinterico non sono alterati.
la sclerosi laterale amiotrofica
In base ai muscoli colpiti per primi e ai segni o sintomi correlati, l’esordio della malattia è classificato come:
- spinale, caratterizzato da un’asimmetria nella forza degli arti. Questo esordio avviene nei tre quarti dei casi colpendo un arto, e influenzando l'andatura e l'abilità manuale: capita, infatti, che i pazienti inciampino spesso o che mostrino difficoltà ad eseguire compiti semplici come abbottonare una camicia, scrivere o girare la chiave nella serratura;
- bulbare, caratterizzato da disartria (disturbo motorio del linguaggio) e disfagia (difficoltà a deglutire), che possono portare all’anartria (completa difficoltà nel pronunciare le parole). Questo esordio riguarda circa un quarto dei casi: Il malato può presentare una certa labilità emotiva con attacchi improvvisi di riso e pianto. In altri casi la malattia si presenta con disfonia, disartria e disfagia per solidi o liquidi ed il successivo coinvolgimento degli arti, nella stragrande maggioranza dei casi, si verifica entro i primi 2 anni. Indipendentemente da quale sia la parte del corpo interessata per prima dalla malattia, la debolezza e l’atrofia muscolare si estendono alle altre parti del corpo. Un esordio bulbare è associato ad una prognosi peggiore rispetto all’esordio spinale.
Questa distinzione di carattere clinico, utile per definire la comparsa della malattia, non appare però sempre così netta nell’evoluzione della stessa, in quanto le due forme possono sovrapporsi.
Dalla diagnosi ai controlli periodici: come si scopre la SLA?
La diagnosi si basa su criteri precisi:
- una degenerazione del motoneurone inferiore
- una degenerazione del motoneurone superiore
- la diffusione progressiva dei sintomi all’interno di una regione o da una regione all’altra.
Il grado di certezza della diagnosi dipende dal numero di distretti corporei colpiti e dal coinvolgimento di entrambi i motoneuroni. Tra l’esordio dei sintomi e la conferma della diagnosi intercorre un periodo di latenza che varia mediamente dai 10 ai 12 mesi.
Oltre agli esami clinici, come supporto diagnostico vengono utilizzati anche:
- l’elettromiografia (EMG), che permette la valutazione del corretto funzionamento dei nervi periferici e dei muscoli
- la Risonanza Magnetica Nucleare (RMN), che consente l’esclusione di altre malattie che coinvolgono il sistema piramidale (sistema nervoso che provvede ai movimenti volontari dei muscoli)
- la PET (Tomografia ad Emissione di Positroni), che consente di studiare il metabolismo di diverse aree cerebrali.
Gli esami clinici devono essere ripetuti per lo meno ogni sei mesi per valutare la progressione della malattia.
Quali sono i medici specialisti coinvolti nella cura di questa patologia?
La SLA è una malattia complessa e progressiva che coinvolge via via diverse funzioni, per cui è necessario l’intervento di diversi specialisti. I centri che si occupano di questa malattia utilizzano un approccio multidisciplinare da parte di:
- Neurologo
- Fisiatra
- Fisioterapista
- Logopedista
- Gastroenterologo
- Pneumologo
- Psicologo
- Genetista
L’epidemiologia della SLA
La Sclerosi Laterale Amiotrofica è una patologia dell’età adulta e può presentarsi in due forme:
- Familiare (5% dei casi), cioè in diversi componenti del nucleo familiare, con esordio intorno ai 63 anni;
- Sporadica (95% dei casi) ossia ad eziologia non nota, con esordio più precoce, tra i 40 e i 60 anni.
In generale vi è una leggera prevalenza del sesso maschile con un rapporto di circa 1,2-1,5.
La prognosi nel 50% dei pazienti è di circa 30 mesi dall’esordio dei sintomi. Il 5-10% dei pazienti sopravvive per più di 8 anni, mentre sono rari i casi in cui si ha una sopravvivenza maggiore. Il decesso si verifica spesso per paralisi della muscolatura volontaria respiratoria.
L’incidenza globale è di 1,7 casi per 100.000 persone/anno, con circa 1000 nuovi casi all’anno in Italia.
La prevalenza globale è attualmente stimata attorno ai 200.000-300.000 casi, circa 5000 in Italia.
Tra i fattori di rischio ambientali per la SLA ci sono:
- i traumi
- il fumo
- l’attività sportiva intensa (leggi articoli correlati)
- l’esposizione ad agenti tossici (es. pesticidi).
Il ruolo di questi fattori non è del tutto definito e non si esclude che l’insorgenza della malattia sia dovuta all’interazione tra fattori genetici ed ambientali.
Le cause genetiche della SLA
La forma familiare ed alcune forme di Sclerosi Laterale Amiotrofica sporadica sono dovute a mutazioni di geni coinvolti in diversi meccanismi fisiopatologici. Oggi quelli più noti sono:
- il gene SOD1, le cui mutazioni sono rappresentate in circa il 20% delle forme familiari e nel 2-3 % di quelle sporadiche. SOD1 è il primo gene scoperto nella SLA e sembra essere associato in maniera esclusiva a questa malattia;
- il geneTARDBP, le cui mutazioni sono presenti solo nel 3% delle forme familiari e nel 1-2% di quelle sporadiche. TDP-43, la proteina codificata da questo gene è presente negli aggregati di proteine che si trovano nei neuroni e nei motoneuroni del 97% dei pazienti con SLA;
- il gene C9orf72, le cui mutazioni sono ad oggi le più rappresentate sia nella SLA familiare (40%) che in quella sporadica (20%) oltre che in alcuni pazienti affetti da demenza frontotemporale. A differenza di altri geni, la mutazione di C9orf72 può provocare sia una perdita di funzione della proteina che un’acquisizione di effetti tossici.
Ad oggi sono più di 30 i geni che presentano un rischio di associazione alla SLA; tuttavia, esiste una ereditarietà che comprende anche altre varianti geniche in cui la malattia si evidenzia solo con la presenza di più di un gene anomalo.
Il test genetico sui soggetti a rischio di malattia familiare viene effettuato nei centri SLA multidisciplinari: qui un consulente genetico aiuta il paziente e i suoi familiari a valutare il rischio e a discutere l'impatto, dal momento che ad oggi non esiste una cura efficace per questa malattia.
Quali sono i meccanismi neuropatologici della SLA?
La caratteristica patologica della Sclerosi Laterale Amiotrofica è la presenza di ammassi di proteine all'interno dei motoneuroni. Questi aggregati sono costituiti per lo più da:
- strutture intracellulari solide chiamate neurofilamenti fosforilati
- una piccola proteina regolatoria chiamata ubiquitina
- la proteina TDP-43 e diverse altre proteine come nelle inclusioni skein-like.
Per questo la SLA viene anche considerata una proteinopatia, analogamente ad altre due malattie neurodegenerative come l’Alzheimer e il Parkinson.
I MODELLI PRECLINICI PER STUDIARE LA SLA
Lo studio dei meccanismi genetici che causano la malattia è stato possibile anche grazie allo sviluppo di modelli preclinici animali, portatori dei geni responsabili della malattia. Tra essi il topo transgenico portatore del gene umano mutato SOD1G93A è quello più studiato per le caratteristiche fenotipiche e neuropatologiche che mimano meglio di altri alcune condizioni dei pazienti SLA. (Figura 3).
Dallo studio di questi modelli si può affermare che la SLA è una patologia multifattoriale e multicellulare. Oltre ai motoneuroni e l’apparato neuromuscolare, la SLA coinvolge anche altri sistemi, come il sistema immunitario e il microbioma intestinale.
Ad oggi le ipotesi che portano all'insorgenza e alla progressione della SLA sono:
- l’alterata omeostasi proteica che provoca i principali meccanismi responsabili della degradazione e della eliminazione di proteine anomale all'interno delle cellule;
- lo stress ossidativo che causa un’eccessiva formazione di radicali liberi tossici;
- la disfunzione dei mitocondri, i principali produttori di energia della cellula;
- l’eccitotossicità, fenomeno provocato da un accumulo di glutammato nei motoneuroni e da una eccessiva stimolazione dei recettori di questo neurotrasmettitore;
- una disfunzione a livello nucleare dei sistema del metabolismo del RNA, dei meccanismi di riparo del DNA e del trasporto nucleo citoplasma
- l’alterazione dei processi di trasporto assonale di vescicole e molecole
- la neuroinfiammazione ossia l’eccessiva attivazione degli astrociti e della microglia
- un'aberrante risposta immunitaria con infiltrati di cellule immunitarie (macrofagi e linfociti) nel midollo spinale, nervi e muscoli.
La Sclerosi Laterale Amiotrofica è ancora oggi purtroppo una malattia incurabile, in quanto nessun trattamento esistente permette al paziente di guarire.
Sebbene siano stati studiati numerosi farmaci con diversi meccanismi d’azione, solo due principi attivi ad oggi hanno ottenuto l’autorizzazione all’immissione in commercio per il trattamento della patologia, ossia il Riluzolo e l’Edaravone.
Riluzolo (Rilutek®) è stato il primo farmaco approvato per la SLA dalla Food and Drug Administration (FDA) negli USA nel 1995, e dall’European Medicine Agency (EMA) l’anno successivo. E’ un antagonista del glutammato il cui meccanismo d’azione non è stato ancora completamente chiarito. Il suo utilizzo permette di posticipare il ricorso alla ventilazione assistita o di prolungare la sopravvivenza mediamente di 3 mesi. La terapia è risultata più efficace se il farmaco viene somministrato a pazienti giovani nel primo stadio della malattia, mentre non ha mostrato efficacia negli stadi più avanzati. Inoltre, non ha mostrato un reale effetto sulla forza muscolare e sui sintomi motori.
Edaravone (Radicava®) approvato dall’FDA nel 2017, è disponibile come soluzione per infusione venosa. Il farmaco ha una potente attività di scovare ed eliminare i radicali liberi. E’ stato dimostrato che l’Edaravone è in grado di rallentare l’aggravamento della malattia in una sottopopolazione di pazienti che riesce ancora a svolgere le normali attività (determina EMA/293450/2019).
Nonostante sino ad oggi siano stati valutati circa altri 60 trattamenti inclusi quelli non-farmacologici (ad esempio, le cellule staminali), nessuno è risultato efficace nel rallentare la malattia.
In assenza di trattamenti efficaci, le terapie sintomatiche e di supporto rimangono la base per la gestione dei pazienti con SLA, e sono:
- trattamenti per il controllo della spasticità
- trattamenti per il controllo dell’ipersalivazione
- trattamenti per il controllo dei crampi muscolari
- trattamenti per il controllo della disfagia e della disartria
- trattamenti per il controllo della trombosi venosa profonda e dell’insufficienza respiratoria.
Molte di queste terapie sintomatiche sono associate ad un chiaro aumento della sopravvivenza, mentre altre alleviano solo in parte i sintomi, portando solo ad un miglioramento della qualità della vita del paziente. È importante notare che la maggior parte delle terapie sintomatiche non è stata testata in studi randomizzati controllati, e si basa dunque sull’esperienza derivante dalla gestione di altre malattie.
Cosa si fa al Mario Negri per la SLA?
Al Mario Negri esiste un gruppo di studio sulla SLA che va dalla ricerca di base preclinica ad approcci traslazionali ed epidemiologici sino al disegno e all'organizzazione di sperimentazioni cliniche. Sebbene l’Istituto non disponga di posti letto per il ricovero dei malati, il gruppo di studio può contare su una vasta collaborazione con diversi centri neurologici in Italia, e non solo, per gli studi traslazionali, epidemiologici e per la sperimentazione di nuovi trattamenti.
La ricerca preclinica è condotta prevalentemente presso il Laboratorio di Neurobiologia Molecolare del Dipartimento di Neuroscienze, coordinato dalla Dr.ssa Caterina Bendotti, e presso il Laboratorio di Biomarcatori Traslazionali del Dipartimento di Biochimica e Farmacologia Molecolare, coordinato dalla Dr.ssa Valentina Bonetto. La ricerca clinica epidemiologica e sperimentale è effettuata presso il Laboratorio di Malattie Neurologiche del Dipartimento di Neuroscienze coordinato dal Dr. Ettore Beghi con la collaborazione della Dr.ssa Elisabetta Pupillo. Attualmente è in atto uno studio clinico frutto della collaborazione fra i tre laboratori: lo studio riguarda l'analisi dell'efficacia del trattamento con RNS60, un composto con azione anti-infiammatoria.
I risultati dei nostri studi preclinici, effettuati in modelli in vitro e in vivo di SLA associati alla mutazione SOD1G93A, dimostrano che il trattamento con RNS60 è in grado di proteggere i motoneuroni e di migliorare la progressione della malattia: i livelli di fattori pro-infiammatori risultano ridotti e la risposta immunitaria a livello periferico del sistema neuromuscolare viene modulata.
A questi risultati è seguito uno studio pilota su 13 pazienti SLA: lo studio ha dimostrato che il trattamento a lungo termine con RNS60 è ben tollerato. Per questo motivo è stato subito attivato uno studio clinico multicentrico su 142 pazienti che ricevono RNS60 o placebo in doppio cieco (Clinical trial NCT03456882), studio che è tuttora in corso . L’obiettivo è quello di valutare l’efficacia del farmaco sulla progressione della malattia e sull’ andamento nel tempo di una serie di biomarcatori nel sangue dei pazienti trattati con RNS60 rispetto a quelli con placebo. I risultati saranno disponibili entro maggio 2021.
Esistono associazioni di pazienti affetti da SLA?

L’associazione italiana per la Sclerosi Laterale Amiotrofica (AISLA) è l’associazione di malati più diffusa sul territorio italiano, a cui afferiscono anche altre associazioni.
La tutela, l’assistenza e la cura dei malati di SLA oltre che la divulgazione di informazioni su questa malattia sono la principale missione di AISLA.

Inoltre esiste la Fondazione Italiana di Ricerca per la Sclerosi Laterale Amiotrofica (AriSLA), un'associazione specifica che ha come scopo primario il finanziamento di progetti di ricerca sulla SLA.