La Sclerosi Laterale Amiotrofica (SLA)
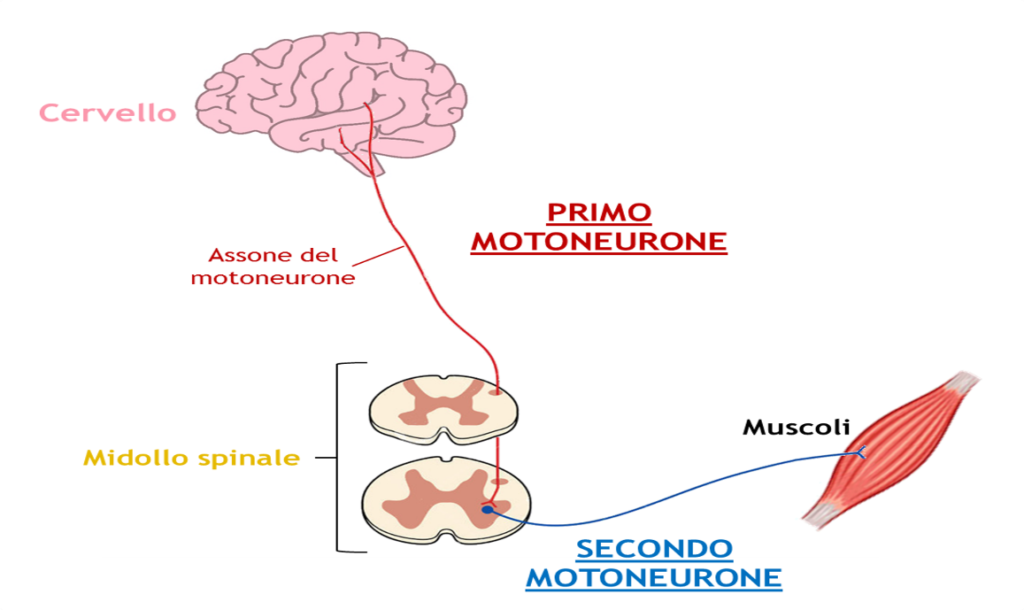
La Sclerosi Laterale Amiotrofica (SLA) colpisce il neurone motorio (o motoneurone), cellula del sistema nervoso centrale. Abbiamo un’atrofia muscolare combinata ad un indurimento di alcune parti del midollo spinale. Si tratta di una patologia degenerativa progressiva che comporta la perdita:
- dei motoneuroni superiori, situati nel cervello e nel tronco encefalico
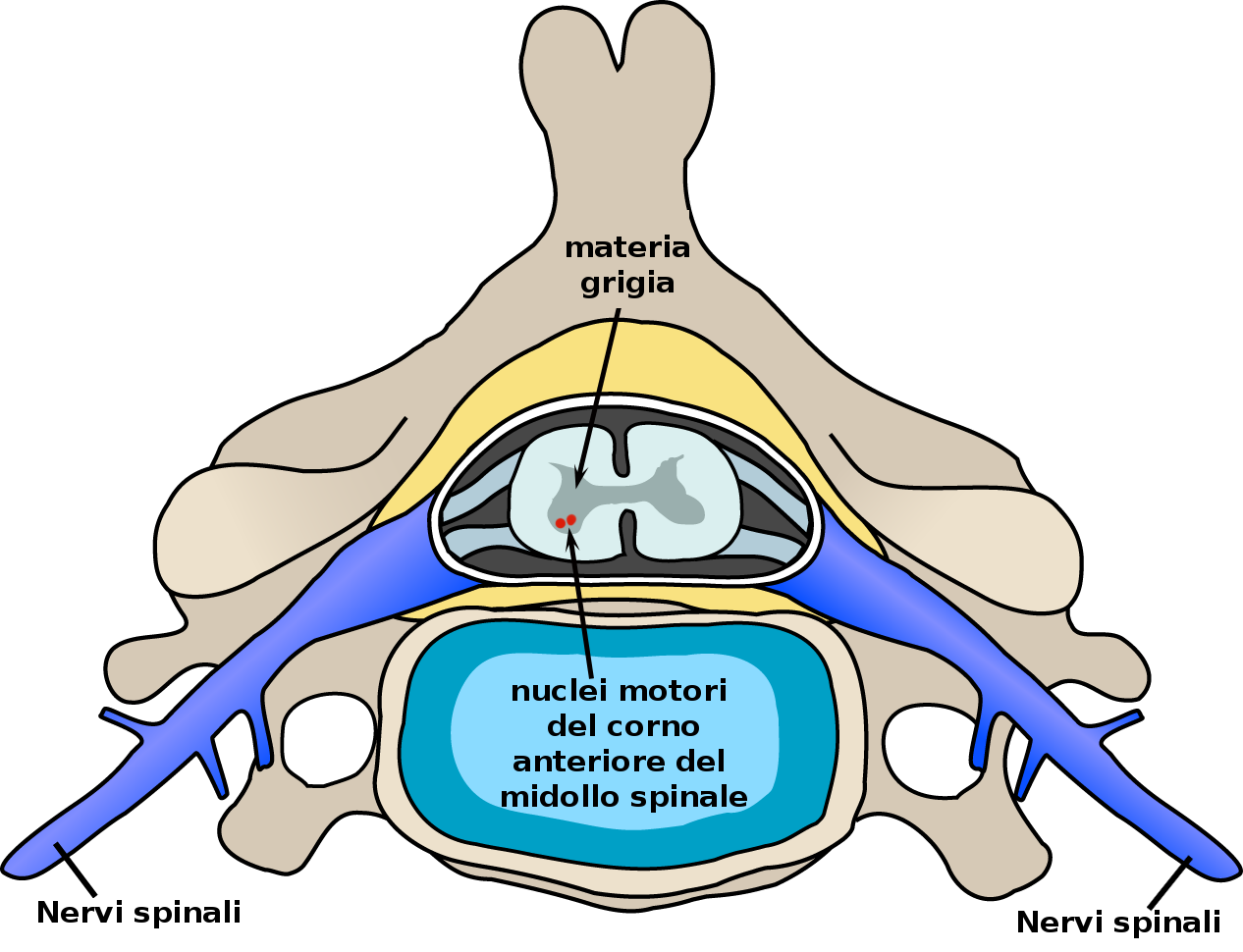
- dei motoneuroni inferiori, situati nel tronco encefalico e nel midollo spinale.
Ciò porta alla mancanza di controllo dei muscoli deputati al movimento e ad altre funzioni. La malattia spesso inizia con spasmi muscolari e debolezza in un arto o difficoltà ad articolare la parola e progredisce sino a colpire tutti i muscoli necessari per muoversi, parlare, mangiare e respirare.
La Sclerosi Laterale Amiotrofica è ancora oggi purtroppo una malattia incurabile, in quanto nessun trattamento esistente permette al paziente di guarire. Nonostante sino ad oggi siano stati valutati circa altri 60 trattamenti inclusi quelli non-farmacologici (ad esempio, le cellule staminali), nessuno è risultato efficace nel rallentare la malattia.
In assenza di trattamenti efficaci, le terapie sintomatiche e di supporto rimangono la base per la gestione dei pazienti con SLA, e sono:
- trattamenti per il controllo della spasticità
- trattamenti per il controllo dell’ipersalivazione
- trattamenti per il controllo dei crampi muscolari
- trattamenti per il controllo della disfagia e della disartria
- trattamenti per il controllo della trombosi venosa profonda e dell’insufficienza respiratoria.
Sintomi
I primi sintomi della malattia possono passare inosservati. Potrebbero manifestarsi inizialmente con delle brevi contrazioni e rigidità muscolare, debolezza dei muscoli fino ad arrivare ad alterato funzionamento di un braccio o di una gamba; voce potrebbe alterata. Questi disturbi (debolezza e atrofia muscolare) piano piano saranno più e si inizierà a sospettare una forma di SLA quando ormai l’entità del danno sarà già considerevole (60-70% di motoneuroni).
I sintomi associati alla degenerazione dei motoneuroni superiori sono:
- aumento eccessivo del tono muscolare (ipertono muscolare)
- accentuazione esagerata dei riflessi muscolo-tendinei (iperreflessia profonda)
- risposta anomala al riflesso cutaneo plantare (segno di Babinski).
I sintomi associati alla perdita dei motoneuroni inferiori sono:
- riduzione del tono muscolare (ipotonia muscolare)
- riduzione del volume muscolare (atrofia muscolare)
- iporeflessia (Diminuzione della capacità di reagire con una risposta nervosa riflessa a determinati stimoli meccanici applicati alla superficie cutanea o a strutture osteotendinee)
- contrazioni muscolari involontarie che si manifestano come dei guizzi visibili sotto la pelle, senza conseguente movimento (fascicolazioni).
Sintomi che si manifestano con il progredire della malattia sono:
- debolezza degli arti;
- crampi muscolari e fascicolazioni;
- difficoltà a camminare o a svolgere le normali attività quotidiane;
- difficoltà a masticare, a deglutire, a parlare e a respirare;
- cambiamenti delle funzioni cognitive e comportamentali.
Di norma il paziente non avverte dolore, tutti gli organi di senso rispondono adeguatamente agli stimoli; la sfera sessuale e il controllo sfinterico non sono alterati.
In base ai muscoli colpiti per primi e ai segni o sintomi correlati, l’esordio della malattia è classificato come:
- spinale: asimmetria nella forza degli arti. Questo esordio avviene nei 3\4 dei casi colpendo un arto, e influenzando l’andatura e l’abilità manuale: capita che s’inciampi spesso o che vi sia difficoltà ad eseguire compiti semplici come scrivere o girare la chiave nella serratura;
- bulbare: disartria (disturbo motorio del linguaggio) e disfagia (difficoltà a deglutire), che possono portare all’anartria (completa difficoltà nel pronunciare le parole). Questo esordio riguarda circa 1\4 dei casi: si può presentare una certa labilità emotiva con attacchi improvvisi di riso e pianto. In altri casi la malattia si presenta con disfonia, disartria e disfagia per solidi o liquidi e successivamente debolezza e l’atrofia muscolare
Diagnosi
La diagnosi si basa su criteri precisi:
- una degenerazione del motoneurone inferiore
- una degenerazione del motoneurone superiore
- la diffusione progressiva dei sintomi all’interno di una regione o da una regione all’altra.
Esami ulteriori:
- l’elettromiografia (EMG), che permette la valutazione del corretto funzionamento dei nervi periferici e dei muscoli
- la Risonanza Magnetica Nucleare (RMN), che consente l’esclusione di altre malattie che coinvolgono il sistema piramidale (sistema nervoso che provvede ai movimenti volontari dei muscoli)
- la PET (Tomografia ad Emissione di Positroni), che consente di studiare il metabolismo di diverse aree cerebrali.
Gli esami clinici devono essere ripetuti per lo meno ogni sei mesi per valutare la progressione della malattia.
Medici specialisti coinvolti
La SLA è una malattia complessa e progressiva che coinvolge via via diverse funzioni, per cui è necessario l’intervento di diversi specialisti: Neurologo – Fisiatra – Fisioterapista – Logopedista – Gastroenterologo – Pneumologo – Psicologo -Genetista
Fattori d’insorgenza e progressione della Sclerosi Laterale Amiotrofica (SLA)
- i traumi
- il fumo
- l’attività sportiva intensa
- l’esposizione ad agenti tossici
- l’alterata omeostasi proteica che provoca i principali meccanismi responsabili della degradazione e della eliminazione di proteine anomale all’interno delle cellule;
- lo stress ossidativo che causa un’eccessiva formazione di radicali liberi tossici;
- la disfunzione dei mitocondri, i principali produttori di energia della cellula;
- l’eccitotossicità, fenomeno provocato da un accumulo di glutammato nei motoneuroni e da una eccessiva stimolazione dei recettori di questo neurotrasmettitore;
- una disfunzione a livello nucleare dei sistema del metabolismo del RNA, dei meccanismi di riparo del DNA e del trasporto nucleo citoplasma
- la neuroinfiammazione ossia l’eccessiva attivazione degli astrociti (sono cellule della glia, i principali costituenti del sistema nervoso insieme ai neuroni. Sono chiamati così perché, visti al microscopio, sono a forma di stella, e svolgono una funzione nutritiva e di sostegno per i neuroni, isolandoli e proteggendoli da lesioni. Uno studio recente rivela senza di essi il nostro orologio biologico sarebbe totalmente sballato) e della microglia (sono un tipo di cellule della glia che si occupano della prima e principale difesa immunitaria attiva nel sistema nervoso centrale (SNC). Sono distribuite, nel cervello e nel midollo spinale, in larghe regioni che non si sovrappongono tra di loro. Le microglia si muovono costantemente e analizzano il SNC in cerca di neuroni danneggiati, placche e agenti infettivi.)
- un’anomala risposta immunitaria che scatena cellule immunitarie come i macrofagi e i linfociti, nel midollo spinale, nervi e muscoli.
Cause genetiche
La forma familiare ed alcune forme di Sclerosi Laterale Amiotrofica sporadica sono dovute a mutazioni di geni coinvolti in diversi meccanismi fisiopatologici. Oggi quelli più noti sono:
- il gene SOD1 è il primo gene scoperto nella SLA e sembra essere associato in maniera esclusiva a questa malattia;
- il gene TARDBP, subisce delle mutazioni; TDP-43, la proteina codificata da questo gene è presente negli aggregati di proteine che si trovano nei neuroni e nei motoneuroni del 97% dei pazienti con SLA;
- il gene C9orf72, le cui mutazioni sono ad oggi le più rappresentate sia nella SLA familiare (40%) che in quella sporadica (20%) oltre che in alcuni pazienti affetti da demenza frontotemporale. Può provocare sia una perdita di funzione della proteina che un’acquisizione di effetti tossici.
L’associazione italiana per la Sclerosi Laterale Amiotrofica AISLA è l’associazione di malati più diffusa sul territorio italiano.
Dott.ssa Logopedista Claudia Antognozzi